
Molecules As Graphs
A cheminformatics team at a contract research organization built a synthesis-cost service. A chemist submits a SMILES string, the service searches for a viable route, estimates reagent and labor cost at the requested scale, and returns a number. Route search is expensive — a few seconds of beam search over a template library, plus calls to external price providers — so the team did the obvious thing and cached results. The cache key was the SMILES string. Submit the same molecule twice, get the second answer instantly.
The cache hit rate was terrible, and nobody could understand why. The same compounds were being requested over and over, and the service kept re-running the full route search anyway. Then someone noticed two entries in the cache that were obviously the same molecule. One chemist had drawn salicylic acid and exported it as OC(=O)c1ccccc1O. Another had drawn the identical structure in a different editor and gotten c1ccccc1(O)C(=O)O. Same atoms, same bonds, same molecule — two different strings. The cache treated them as two different compounds, ran the route search twice, and worse, returned two slightly different cost estimates because the two searches explored the template space in a different order. A customer who happened to submit both spellings got two prices for one molecule and reasonably concluded the service was broken.
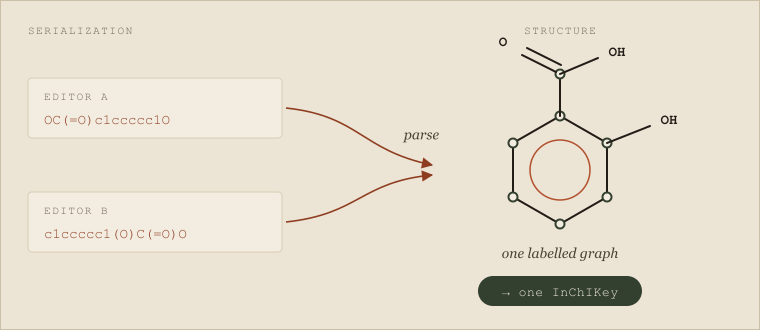
The same molecule, written two ways. Both SMILES strings serialize one labelled graph of salicylic acid; canonicalization collapses them to a single key, so a cache keyed on identity — not on the string — hits once and returns one price.
The bug was not in the pricing code, the route search, or the cache. It was a category error one level below all of them. The team had used the SMILES string as the identity of the molecule, when the identity of a molecule does not live in any particular string. A molecule has many valid SMILES spellings — the notation records one depth-first walk over the structure, and there are as many walks as there are starting atoms and branch orderings. The thing that is actually the same across all those spellings is the graph: the set of atoms and the bonds between them. The string is a serialization of the graph. Keying a cache on the serialization instead of the structure is like keying a cache of people on the order you happened to read their names.
This is a small bug with an expensive lesson, and it is a single instance of the most important idea in computational chemistry. The reason a laptop can search a billion-compound library, plan a synthesis, or flag a structural alert in milliseconds is that we represent molecules as graphs — atoms as nodes, bonds as edges — and chemical questions as graph algorithms. That abstraction is what makes the field tractable. It is also the source of its most subtle and most expensive failures, because a molecule is not only a graph, and the information the graph quietly discards has a way of coming back to matter at exactly the wrong moment. This article is about both halves of that statement: why the graph abstraction is so productive, and where it leaks.
The Abstraction: Atoms as Nodes, Bonds as Edges
A graph, in the mathematical sense, is a set of nodes (or vertices) and a set of edges connecting pairs of them. That is the entire definition. Graphs are studied because an enormous range of structures — road networks, social connections, circuit diagrams, dependency trees — reduce to "things and the relationships between them," and once you have made that reduction, a deep and general body of algorithms becomes available.
A molecule reduces to a graph with almost no friction. The atoms are the nodes. The covalent bonds are the edges. The fit is so natural that it predates computers entirely: the structural formulas chemists have drawn since August Kekule and Archibald Couper in the 1850s are graph drawings, with letters for nodes and lines for edges. When cheminformatics arrived a century later, it did not invent a representation so much as formalize the one chemists were already using.
What makes the molecular graph powerful rather than merely tidy is that the nodes and edges are labeled, and the labels carry chemistry. A node is not an abstract point; it is an atom with an element (carbon, nitrogen, oxygen), a formal charge, a count of attached hydrogens, and — once the graph's cycles are computed — a fact about whether it sits in a ring. An edge is not a bare connection; it is a bond with an order: single, double, triple. A labeled graph of this kind captures essentially the whole of what a chemist means by "constitution" — which atoms are present and how they are connected — and constitution is what most chemical questions are actually about.
Concretely, this is a data structure. In MolBuilder, the molecular-engineering toolkit developed at Wedge, the core object is a Molecule that stores atoms with their element and charge, an explicit list of bonds with their orders, and an adjacency structure that answers the one question every graph algorithm asks first: given this atom, what is it bonded to? A method as humble as neighbors(i) — return the indices of the atoms bonded to atom i — is the primitive on which everything else is built. Ring perception, canonical numbering, fingerprinting, substructure matching: each of them is, underneath, a disciplined way of walking the adjacency structure. The representation is not glamorous. Its productivity comes precisely from being a faithful, minimal encoding of connectivity onto which the machinery of graph theory drops without modification.
One detail of that encoding is worth pausing on, because it is the first place the graph quietly compresses reality and the first convention a newcomer trips over. Most cheminformatics representations do not store hydrogen atoms as explicit nodes. They store the heavy atoms — carbon, nitrogen, oxygen, and the rest — and attach to each a hydrogen count, an integer label saying how many hydrogens hang off it. Ethanol, which has nine atoms, is stored as a three-node graph (two carbons and an oxygen) with hydrogen counts of three, two, and one. This "suppressed-hydrogen" graph is far smaller and far faster to work with, and it is justified by a valence model: given an atom's element, charge, and bonds to heavy atoms, the number of hydrogens needed to complete its normal valence is usually inferable, so storing it explicitly would be redundant. The compression is real leverage — it shrinks the graph by more than half for a typical organic molecule — but it is also the first leak in miniature, because "usually inferable" is not "always inferable." Radicals, unusual oxidation states, and elements with multiple common valences break the inference, and a system that assumes it can always reconstruct the hydrogens will silently miscount them. The lesson recurs at every level of this article: each compression that makes the graph tractable also encodes an assumption that some molecule, somewhere, will violate.
The rest of this article walks through four chemical questions, each of which becomes a classical graph problem the moment you accept the abstraction — identity, similarity, substructure, and reaction — and then turns to the question the abstraction cannot answer, which is where the trouble lives.
Identity: Are These the Same Molecule?
The cache bug is, formally, a question about graph isomorphism. Two graphs are isomorphic if there is a one-to-one correspondence between their nodes that preserves the edges — if you can relabel the atoms of one to make it identical to the other. "Are these two SMILES the same molecule?" is exactly "are these two labeled graphs isomorphic?"
In full generality, graph isomorphism is a famously hard problem. It is not known to be solvable in polynomial time, and it is not known to be NP-complete either; it occupies a strange intermediate status that has fascinated complexity theorists for decades. In 2016 Laszlo Babai proved that it can be solved in quasipolynomial time, a landmark result, but quasipolynomial is still not the kind of bound you want in the inner loop of a database that ingests millions of structures a day.
Molecular graphs, fortunately, are not arbitrary graphs. They are small, sparse, and richly labeled. Carbon has at most four bonds; the elements are few; the node labels (element, charge, hydrogen count) immediately rule out most candidate correspondences before any graph search begins. In practice, deciding whether two molecules are the same is cheap. But "cheap to decide for one pair" is not the same as "cheap to organize a database around." If you have a hundred million compounds and a new structure arrives, you cannot run a pairwise isomorphism test against all hundred million to ask "have I seen this before?" You need each molecule to carry an identity you can look up directly — a key.
This is the problem canonicalization solves. The idea is to compute, for any molecule, a single canonical form that is identical for all of its isomorphic spellings and different for everything else — a fingerprint of identity, not of similarity. If you can do that, identity lookup collapses from a graph search to a string comparison, and the entire apparatus of hash tables and database indexes becomes available.
The foundational technique is Morgan's algorithm, published by H. L. Morgan at Chemical Abstracts Service in 1965 — one of the genuinely seminal papers of the field. Morgan's insight was to assign each atom an "extended connectivity" value computed iteratively: start with each atom's degree (its number of neighbors), then repeatedly replace each atom's value with the sum of its neighbors' values, and continue until the number of distinct values stops growing. Atoms in different structural environments accumulate different values, and those values induce a canonical ordering of the atoms. Once the atoms are canonically ordered, you can write the structure down in exactly one way.
It is worth running the first step by hand on a small molecule, because the mechanism is more intuitive than the description. Take isobutane: a central carbon bonded to three methyl carbons. In round zero, label each atom with its degree — the central carbon gets 3, the three outer carbons get 1 each. In round one, replace each atom's label with the sum of its neighbors' labels: the central carbon, whose three neighbors each carry 1, becomes 3; each outer carbon, whose single neighbor carries 3, becomes 3. Now everything reads 3, which is uninformative, so the rule is to keep iterating only while the count of distinct values increases, and to stop when it stops growing. The values at the last informative round rank the atoms, the central carbon distinguished from the periphery, and that ranking is the seed of a canonical numbering. The reason real canonicalization is more involved than this sketch is symmetry: in a molecule with genuine topological symmetry, several atoms are truly equivalent and Morgan's values tie. Ties must be broken with additional invariants — element, charge, bond orders, chirality — applied in a fixed priority order, which is exactly what Weininger's canonical-SMILES generation algorithm (CANGEN) systematized in 1989 to give every molecule a unique SMILES string. The IUPAC InChI standard, introduced in 2005 and formalized in the literature in 2015, took the idea further into a layered, openly specified identifier, with the hashed InChIKey serving as a fixed-length database key. The InChIKey is a hash, which means it is not collision-proof in principle — two different molecules could in theory share a key — but the space is large enough that a collision has the practical status of a meteor strike, and the convenience of a fixed-length, directly indexable identifier is worth the theoretical risk.
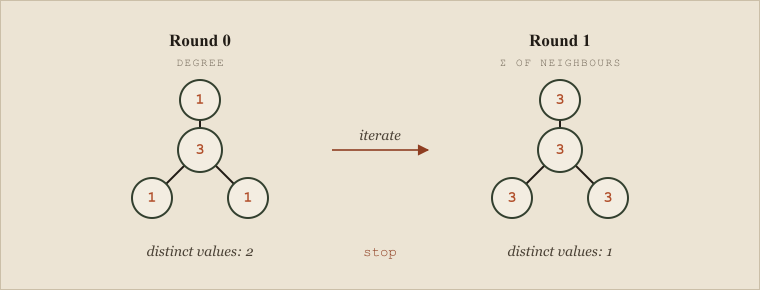
Morgan's extended connectivity on isobutane. Each atom starts labelled by its degree, then takes the sum of its neighbours' labels; when the count of distinct values stops growing (2 -> 1) iteration halts and the central carbon is set apart from the three equivalent methyls. Ties like this are what canonical SMILES (CANGEN) breaks with further invariants.
The practical lesson is the one the CRO team learned the hard way: identity must be computed, not assumed. MolBuilder's own canonicalization module makes the point explicitly in its design. Its core SMILES writer is deliberately not canonical — different depth-first traversal orders produce different valid strings for the same molecule — so the toolkit provides a separate canonicalization step whose entire job is to be the single source of truth for compound identity. It computes an order-invariant canonical SMILES with its own Morgan-based writer — no external dependency required — and, when the cheminformatics ecosystem's standard libraries are available, additionally emits an InChIKey as the strongest identifier; the price cache, the catalog lookup, and the crowd-quote table all key on that canonical identifier rather than on whatever string a caller happened to pass. Salicylic acid written two ways resolves to one key, hits one cache entry, and returns one price. The fix for the opening bug is not cleverer caching. It is recognizing that the string was never the identity in the first place.
Similarity: How Close Are Two Molecules?
Identity is a yes-or-no question. Similarity is the graded version, and it is the question that drives most of practical drug discovery: given a molecule with the activity you want, find others that are like it, on the well-supported assumption that similar structures tend to have similar properties. Turning "alike" into a number that a computer can rank a million compounds by is the job of the molecular fingerprint.
The dominant family of fingerprints is circular, or Morgan, fingerprints — the same Morgan whose extended-connectivity idea solved canonicalization, reused for a different purpose. The modern formulation, extended-connectivity fingerprints (ECFP), was specified by David Rogers and Mathew Hahn in 2010, and the algorithm is worth stating because its structure echoes through everything that follows. Begin by assigning each atom an initial identifier that hashes together its local invariants: element, charge, degree, attached-hydrogen count, ring membership. Then iterate. On each round, replace each atom's identifier with a hash of its own identifier together with the identifiers of its neighbors and the orders of the connecting bonds. After one round, each atom's identifier encodes its immediate neighborhood; after two rounds, the neighborhood two bonds out; after three, three bonds out. The fingerprint is the set of all identifiers seen across all rounds, usually folded down into a fixed-length bit vector. Each bit asserts the presence of some local structural pattern at some radius.
The set of identifiers a molecule generates this way can be large, so it is almost always folded down into a fixed-length bit vector — say 2,048 bits — by taking each identifier modulo the vector length and setting that bit. Folding is what makes fingerprints uniformly sized and fast to compare, but it introduces a small, deliberate sloppiness: two genuinely different substructural features can hash to the same bit, a collision that makes two molecules look marginally more similar than they are. Larger vectors collide less and cost more memory; the standard sizes are an engineering compromise, not a law of nature. The radius of the iteration is the other knob, and it has a chemical meaning. A radius of two — the common default, often written ECFP4 because it spans a diameter of four bonds — captures features up to two bonds from each atom, roughly the size of a functional group and its immediate context. Push the radius higher and the fingerprint encodes larger, more specific fragments, which sharpens discrimination but blurs the very similarity you were trying to detect, because larger fragments are shared by fewer molecules.
MolBuilder implements exactly this — ECFP-style circular fingerprints built from scratch, following Rogers and Hahn, folded into a bit vector — and uses them to score how alike two molecules are and to drive "find me something like this" screening. That is a different question from identity, and the toolkit keeps them separate: exact-duplicate detection keys on the canonical form of the previous section, not on fingerprint overlap, because two molecules can share most of their bits and still be different compounds. The similarity of two fingerprints is measured with the Tanimoto coefficient: the number of bits the two molecules share divided by the number of bits either one has. It runs from zero (no patterns in common) to one (identical pattern sets). Practitioners carry rough thresholds — a Tanimoto of 0.85 or above is "highly similar," typically a shared scaffold; 0.7 is "related"; below 0.3 is "different" — and those heuristics are useful precisely because they are coarse. They should never be mistaken for a metric of biological similarity: two molecules can be 0.9 Tanimoto-similar and differ by the one substituent that abolishes binding, a phenomenon medicinal chemists call an activity cliff, and the fingerprint, by construction, cannot see the cliff coming.
There is a deeper reason to understand circular fingerprints than knowing how to call the function, and it is the most beautiful connection in this entire subject. That iterative scheme — each atom repeatedly absorbing a summary of its neighbors' states — is not unique to chemistry. It is the Weisfeiler-Leman algorithm, a graph-isomorphism heuristic published by Boris Weisfeiler and Andrei Leman in 1968. WL refines node labels by iterated neighborhood aggregation in order to tell graphs apart, which is precisely what Morgan's extended connectivity does and precisely what ECFP does. The three were invented independently, for canonicalization, for similarity, and for isomorphism testing, and they are the same algorithm.
And that same algorithm is the conceptual core of the graph neural networks that now dominate molecular machine learning. A message-passing neural network — the framework Justin Gilmer and colleagues unified in 2017 — has each atom repeatedly receive "messages" from its neighbors, aggregate them, and update its own state, for a fixed number of rounds, after which the atom states are pooled into a molecule-level representation. That is Weisfeiler-Leman with the fixed hash function replaced by a learned neural network. The connection is not a loose analogy; Keyulu Xu and colleagues proved in 2019 that a message-passing network is, at most, exactly as powerful at distinguishing graphs as the WL test it generalizes. The hand-engineered fingerprint your similarity search uses and the deep network your property model uses are the same idea at different temperatures — one with a frozen hash, one with learned weights.

The same procedure wearing four hats. Morgan's canonicalization, the ECFP fingerprint, the Weisfeiler-Leman isomorphism test, and a graph neural network all iterate the same neighbourhood aggregation — only the hash differs: fixed in the first three, learned in the fourth. They share the same blind spots.
Knowing that they are the same algorithm tells you something important and practical about both: a graph neural network inherits Weisfeiler-Leman's blind spots, and those blind spots are not abstract. WL, and therefore standard message passing, cannot reliably count rings or distinguish certain symmetric ring systems, because the iterated neighborhood aggregation that gives each atom a summary of its surroundings cannot always tell a six-membered ring from two fused smaller ones when the local environments happen to look identical. The textbook illustration is that a single large ring and a pair of separate smaller rings with the same total atom count can produce identical WL labels, and a no-frills GNN will assign them the same representation. For molecules, where ring size and ring fusion are first-order determinants of shape and reactivity, that is a real limitation, and it is why a serious property model usually augments raw message passing with explicit ring and substructure features rather than trusting the network to rediscover them. The lesson is the recurring one of this article in a new guise: the most powerful tool in the box is still a graph algorithm, and it inherits the graph's silences. There are non-isomorphic molecules that no amount of message passing can tell apart, just as there are distinct molecules with identical ECFP fingerprints, and knowing which ones is the difference between a model you trust and a model you merely hope about.
Substructure: Does This Contain That?
A great deal of chemistry is reasoning about parts. Does this molecule contain a carboxylic acid? A nitro group on an aromatic ring? A reactive Michael acceptor that should keep it out of a screening collection? Each of these is a question about whether a small pattern graph appears somewhere inside a larger molecular graph — the subgraph isomorphism problem.
Subgraph isomorphism is genuinely hard: in the general case it is NP-complete, one of the canonical intractable problems of computer science. Unlike the full-graph isomorphism of the identity question, there is no quasipolynomial reprieve here. What rescues it in practice is, once again, that molecular graphs are small and richly labeled, and that decades of engineering have gone into the search. J. R. Ullmann's 1976 backtracking algorithm remains the textbook foundation; the VF2 algorithm of Cordella and colleagues, published in 2004, is the workhorse behind most modern substructure search, using carefully ordered feasibility rules to prune the search tree before it explodes. Most cheminformatics toolkits, MolBuilder included, implement a labeled subgraph matcher of this lineage.
The query side of substructure search has its own language: SMARTS, an extension of SMILES designed not to name a specific molecule but to describe a pattern to match. Where SMILES says "an aromatic carbon," SMARTS can say "an aromatic carbon that is in a six-membered ring and bonded to a halogen." A SMARTS pattern is a small labeled graph with logical conditions on its nodes and edges — a node can demand a particular element or any of a set of elements, a specific number of connections, a ring size, a charge — and matching it against a molecule is subgraph isomorphism with predicate tests at each node. The language is expressive enough to be genuinely a query language rather than a fixed pattern: recursive SMARTS lets a node's condition itself be a whole sub-pattern ("a carbon that is attached to a carbonyl that is attached to an aromatic ring"), so that the definition of a functional group can encode the environment that makes it reactive rather than just its bare atoms. That expressiveness is exactly why structural alerts can be written precisely. MolBuilder's SMARTS engine — a tokenizer, parser, and matcher — underlies its structural-alert filters (the PAINS-style and toxicophore patterns that flag compounds likely to misbehave in assays or in the body), its controlled-substance screen, and its reaction templates, which we come to next; it also cross-validates the toolkit's faster heuristic functional-group detector, which does the routine detection by walking the graph directly.
The operational catch is scale. Running a VF2 match against one molecule is fast; running it against a billion-compound catalog, one molecule at a time, to answer "which compounds contain this scaffold?" is not. The standard remedy is a two-stage filter that reuses the fingerprint machinery from the previous section. A screening fingerprint records, for each molecule, the presence of a fixed set of small substructural features. The logic rests on a simple monotonicity: if a query pattern is genuinely contained in a candidate molecule, then every feature of the query must also be present in the candidate, so the query's fingerprint bits must be a subset of the candidate's. The contrapositive is what does the work — if the query has any bit the candidate lacks, the candidate cannot contain the query, and it can be rejected by a single bitwise test (does query AND candidate still equal query?) without ever invoking the graph search. The fingerprint pre-filter discards the overwhelming majority of non-matches in microseconds, and the exact subgraph isomorphism test runs only on the small surviving set of candidates that passed the subset check. The pre-filter is allowed false positives — molecules that pass the bit test but fail the real match — but never false negatives, which is the only guarantee that matters for correctness. This is the same pattern that makes every other billion-scale operation feasible: an approximate, cheap structural summary, designed to be conservative in the safe direction, sitting in front of an exact and expensive graph algorithm.
Reactions as Graph Edits
If a molecule is a graph, a reaction is a graph edit. A reaction takes one set of molecular graphs (the reactants) and produces another (the products) by breaking some bonds, forming others, and occasionally adding or removing atoms — a transformation of the edge set, with the node set largely conserved. The bookkeeping that tracks which reactant atom becomes which product atom is called atom mapping, and it is precisely a partial isomorphism between the reactant and product graphs.
This reframing is what makes computer-assisted synthesis planning possible. A reaction template is a subgraph rewrite rule: a SMARTS-like pattern that says "wherever you find this arrangement of atoms and bonds, you may replace it with that arrangement." Run a template forward and you predict a product; run it backward — retrosynthesis — and you ask "what simpler precursors, under this transformation, would yield my target?" MolBuilder's retrosynthesis engine works exactly this way, applying a library of reaction templates through a beam search that explores the tree of possible disconnections, scoring each route by feasibility and recursing on the precursors until it reaches purchasable starting materials.
The hard part, and the place the graph view earns its keep, is atom mapping. To extract a reaction template from a recorded reaction — or to apply one cleanly — you must know which atom in the product came from which atom in the reactant, a correspondence that experimental data almost never records. Recovering it is, formally, finding the maximum common substructure between reactant and product graphs: the largest subgraph they share, which is the part the reaction left untouched, with the bond changes localized to its boundary. Maximum common substructure is itself an NP-hard graph problem, and getting atom mapping wrong produces templates that are subtly malformed — they match the wrong contexts and propose impossible disconnections — which is one quiet reason template-based route planners vary so much in quality. The 2018 demonstration by Marwin Segler and colleagues that deep neural networks combined with symbolic template application could plan syntheses competitive with human chemists is, underneath the machine learning, an enormous exercise in graph rewriting guided by learned priors, and the quality of the underlying atom-mapped templates is as much the foundation as the neural network on top of it.
The point for this article is not the mechanics of route search but the continuity of the abstraction. The same graph that gave us identity, similarity, and substructure also gives us reactivity, because templates are subgraph patterns and reactions are subgraph rewrites. A single representation carries the molecule from "what is it" through "what is it like" and "what does it contain" all the way to "how do you make it." That continuity is the quiet reason a single toolkit can span from a parsed structure to a manufacturing cost estimate without ever changing its underlying model of what a molecule is.
Where the Abstraction Leaks
Everything above is the case for the prosecution's opposite — a celebration of how much chemistry collapses cleanly onto graph theory. Now the necessary correction. A molecule is not only a graph, and the gap between "molecule" and "labeled graph of atoms and bonds" is where the field's most expensive and most embarrassing bugs are born. These are not exotic edge cases. They are everyday chemistry that the bare abstraction cannot represent, and every one of them has, at some point, silently corrupted a database, mismatched a compound, or invalidated a model.
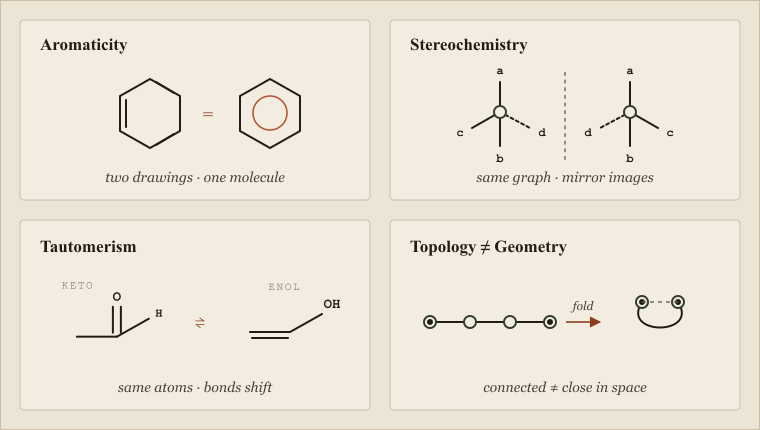
Four leaks. Aromaticity: one molecule, two equally valid drawings. Stereochemistry: identical connectivity, non-superimposable mirror images. Tautomerism: the same atoms with a hydrogen and a double bond moved. Topology vs. geometry: a connected chain folds so atoms far apart in the graph become neighbours in space.
Aromaticity is the first leak, and it begins with a question that sounds like it should have an answer: what are the bonds in benzene? Drawn as a graph in the most literal way, benzene is a six-membered ring of alternating single and double bonds — a Kekule structure. But there are two equivalent Kekule structures, and the real molecule is neither; its six carbon-carbon bonds are identical, the pi electrons delocalized around the ring. Cheminformatics toolkits handle this by introducing an "aromatic" bond type and a model that decides which atoms and bonds count as aromatic — and crucially, different toolkits use different aromaticity models. One system's aromatic perception may flag a ring that another's does not. A molecule canonicalized under one model and matched under another can fail to match itself. The leak is that aromaticity is a fact about electronic structure that has been forced onto a representation built for localized bonds, and the patch — an extra bond type plus a perception model — is not standardized across the ecosystem.
Stereochemistry is the second, and it is the one that has killed people. A graph encodes which atoms are bonded to which; it says nothing about their arrangement in space. But two molecules can have identical connectivity and be non-superimposable mirror images — enantiomers — with dramatically different biological behavior. The bare graph cannot tell them apart, because as graphs they are isomorphic. Cheminformatics bolts stereochemistry on with annotations: parity flags on tetrahedral centers for R/S configuration, directional markers on double bonds for E/Z geometry. These annotations are real and necessary, but they are exactly that — annotations layered on top of a representation that does not natively carry them, easy to drop in a file-format conversion, easy to mismatch, easy to ignore in a fingerprint that was only ever computed over the connectivity graph. The cautionary tale every cheminformatician knows is thalidomide, whose two enantiomers differed in connectivity not at all and in effect catastrophically. A system that treats the molecular graph as the whole molecule treats enantiomers as one compound, and there are contexts in which that is a fatal error rather than a rounding one.
Tautomerism is the third and the most philosophically unsettling, because it makes even the identity question ambiguous. A tautomer pair — the keto and enol forms of a carbonyl compound are the classic example — consists of two structures that interconvert by moving a hydrogen and shifting a double bond. As graphs they are genuinely different: different bonds, different hydrogen positions. As chemistry they are the same substance in dynamic equilibrium, and which form you draw is often arbitrary. So: are two tautomers the same molecule or not? There is no clean answer, and the consequences are practical. A database that canonicalizes tautomers to a single form will unify them; one that does not will store them separately, and a search for one will miss the other. As Wendy Warr documented in a 2010 survey of the problem, tautomer handling is one of the genuine swamps of chemical information management, and it is a swamp precisely because the graph abstraction insists on a single definite structure while the chemistry refuses to provide one.
Charge and resonance leak in a related way. Where, exactly, is the double bond in a carboxylate anion? The two oxygens are equivalent, the negative charge and the pi bond delocalized between them, but a graph with integer bond orders and integer formal charges must commit to drawing the double bond to one oxygen and the negative charge on the other. The representation forces a localized fiction onto a delocalized reality. Most of the time it does not matter; occasionally, when a fingerprint or a template match depends on which oxygen got the double bond, it matters a great deal.
The integer-bond-order model leaks further still at the edges of organic chemistry. The whole scheme assumes a bond is a shared pair of electrons that can be counted as one, two, or three — and for the carbon-rich molecules of drug discovery, that assumption holds beautifully. It breaks for organometallic compounds, where a metal center may be coordinated by a whole aromatic ring through a bond that belongs to no integer order, and for dative or coordinate bonds, where both electrons come from one partner. Toolkits paper over this with conventions — a special "dative" bond type, or simply drawing the metal as disconnected and relying on charges — and, predictably, the conventions are not standardized, so the same metal complex can be represented several incompatible ways and fail to match itself across systems. A related, smaller leak is isotopic labeling: a deuterium and a protium are the same node as far as the unlabeled graph is concerned, yet they are different for the purposes of a mass-spec study or a metabolically stabilized drug, and the distinction survives only as another optional annotation that a connectivity-only fingerprint will ignore. None of these cases is common in a typical medicinal-chemistry library, which is exactly why they are dangerous: they are rare enough to be untested and real enough to corrupt the one project that hits them.
Ring perception is a subtler leak, and an instructive one because here the chemistry is unambiguous but the graph theory is not. Chemists speak freely of "the rings" in a molecule, and for a simple structure the rings are obvious. But a fused or bridged polycyclic system — the steroid nucleus, or a cage like cubane or norbornane — does not have a single well-defined set of rings as a matter of graph theory. The space of cycles in such a graph is large, and the convention most toolkits adopt, the smallest set of smallest rings (SSSR), is not even unique: some molecules admit several equally valid smallest-ring sets, and the algorithm has to pick one. Because ring membership feeds aromaticity perception, fingerprint invariants, and SMARTS ring-size predicates, two toolkits that resolve the same ambiguous cage differently can disagree all the way up the stack, on a molecule where no chemist would say there was anything to disagree about. The leak here is not that the graph discards information; it is that a chemical intuition ("the rings") maps onto no single graph-theoretic object, and the gap is filled by a convention that different implementations fill differently.
The deepest leak of all is that a graph encodes topology, not geometry. Connectivity is not shape. Two atoms five bonds apart along the chain may be neighbors in space when the molecule folds; a binding event depends on the three-dimensional arrangement of atoms presented to a protein pocket, not on the bond-counting distance between them. The molecular graph is silent about conformation, about distance, about the actual shape that does the chemistry of recognition. MolBuilder, like any serious toolkit, maintains separate machinery for this — conformer generation, internal-coordinate geometry, the explicit 3D structure — precisely because the graph cannot supply it. This is the same lesson, viewed from the structural side, that we reached in the article on representing molecular interaction data: every representation encodes some questions cleanly and lets others fall away, and the graph's blind spot is geometry. When the question is "is this the same compound?" or "what does this contain?", the graph is the right tool and shape is a distraction. When the question is "will this bind?", the graph has thrown away the very information the question is about.
Working With the Leak, Not Against It
None of this is an argument against the graph abstraction. It is the foundation of the field for good reason, and nothing proposed here replaces it. The argument is narrower and more useful: an engineer who knows where a model leaks builds systems that survive contact with real data, and one who mistakes the model for the molecule builds systems that fail in ways that are maddening to diagnose because the code is all correct. A handful of disciplines follow directly from taking the leaks seriously.
Canonicalize at the boundary. Any time a molecule enters your system — a user submission, a file import, an API call — compute its canonical identity immediately and key everything internal on that, never on the raw string you were handed. The opening bug disappears entirely under this one rule, and so do a large fraction of the deduplication and cache-coherence problems that plague chemical databases. Use the strongest identifier your installation supports; an InChIKey is a better key than a canonical SMILES, which is a far better key than the string a user typed.
Match the representation to the question. The graph is the right model for identity, similarity, and substructure, and the wrong model for anything that depends on shape. If your task is binding-affinity prediction or pharmacophore matching, the graph is a starting point, not an endpoint, and you need the 3D layer. Reaching for a graph fingerprint because it is what your library makes easy, when the question is fundamentally geometric, is the representational equivalent of looking for your keys under the streetlight.
Know what your fingerprint preserves and what it discards. A circular fingerprint computed over the connectivity graph is, by construction, blind to stereochemistry unless you have explicitly included stereo information in the atom invariants. If your similarity search treats enantiomers as identical and your application cares about chirality, you have a silent correctness bug, not a tuning parameter. The fix is to know the contents of your representation precisely enough to state what it cannot distinguish.
Test at the seams. The bugs are not in the middle of the abstraction; they are at its edges. A test suite for a cheminformatics system earns its keep by covering exactly the leaks: a tautomer pair that must (or must not) unify, an enantiomer pair that must stay distinct, an aromatic ring that must match across perception models, a fused polycyclic cage whose ring set is ambiguous, a charged species with delocalized bonds, a molecule whose folded conformation matters. These cases are where systems quietly diverge from chemistry, and a handful of them in a test file is worth more than a thousand assertions about molecules that never stress the model.
Treat the canonicalizer and the fingerprint definition as versioned dependencies. This one is easy to miss because it does not look like a leak; it looks like an upgrade. The canonical identity of a molecule and the bits of its fingerprint are not platonic facts — they are outputs of a specific algorithm in a specific version of a specific toolkit. Change the aromaticity model, refine the tie-breaking in canonicalization, or alter the fingerprint's atom invariants between releases, and the same molecule can acquire a different canonical key or a different bit vector than it had last year. A database keyed on last year's canonicalizer and queried with this year's will miss compounds it already contains, and a model trained on one fingerprint version and served another will degrade silently. The discipline is to record which version produced every stored identifier and fingerprint, and to recompute deliberately rather than letting a dependency bump quietly redefine identity underneath you.
Underneath all four is one habit of mind. A representation is a model, and every model has a domain of validity outside which its answers are not wrong so much as meaningless. The graph model of molecules is extraordinarily wide in its domain — it is the reason most of computational chemistry exists — but it is not infinite, and the boundary is not where the math gets hard. It is where the chemistry stops being about connectivity and starts being about electrons, mirror images, equilibria, and shape.
The Information You Threw Away
The molecular graph is the most productive abstraction in chemistry because of what it throws away, not in spite of it. By discarding geometry, electronic delocalization, and dynamics, and keeping only atoms and the bonds between them, it reduces the bewildering richness of a real molecule to a structure that decades of graph theory already knew how to handle. Identity becomes isomorphism. Similarity becomes fingerprint overlap. Substructure becomes subgraph matching. Synthesis becomes graph rewriting. A laptop can do in milliseconds what the abstraction makes cheap, and almost everything in cheminformatics — the searchable databases, the property models, the retrosynthesis engines, the structural alerts — is built on the leverage that one reduction provides.
And the failures of the field are, with remarkable consistency, the discarded information coming back to be paid for. The cache that quoted two prices for one molecule, the search that missed a tautomer, the model that confused two enantiomers, the match that failed across two aromaticity perceptions — each is the graph abstraction working exactly as designed, faithfully ignoring something that turned out to matter. The information was not lost by accident. It was thrown away on purpose, because throwing it away is what made the rest tractable, and the engineer's job is to remember what was thrown away and to put it back precisely where, and only where, the question demands it.
A molecule is a graph. That sentence is true enough to build a field on and false enough to break a system that believes it completely. Holding both halves at once — using the abstraction for everything it is good for, and knowing exactly where it stops describing the molecule — is not a footnote to computational chemistry. It is the craft itself.
References
Morgan, H. L. (1965). The generation of a unique machine description for chemical structures — a technique developed at Chemical Abstracts Service. Journal of Chemical Documentation, 5(2), 107-113. doi:10.1021/c160017a018
Weininger, D. (1988). SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of Chemical Information and Computer Sciences, 28(1), 31-36. doi:10.1021/ci00057a005
Weininger, D., Weininger, A., & Weininger, J. L. (1989). SMILES. 2. Algorithm for generation of unique SMILES notation. Journal of Chemical Information and Computer Sciences, 29(2), 97-101. doi:10.1021/ci00062a008
Heller, S. R., McNaught, A., Pletnev, I., Stein, S., & Tchekhovskoi, D. (2015). InChI, the IUPAC International Chemical Identifier. Journal of Cheminformatics, 7, 23. doi:10.1186/s13321-015-0068-4
Rogers, D., & Hahn, M. (2010). Extended-connectivity fingerprints. Journal of Chemical Information and Modeling, 50(5), 742-754. doi:10.1021/ci100050t
Weisfeiler, B., & Leman, A. (1968). The reduction of a graph to canonical form and the algebra which appears therein. Nauchno-Technicheskaya Informatsia, 2(9), 12-16.
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O., & Dahl, G. E. (2017). Neural message passing for quantum chemistry. Proceedings of the 34th International Conference on Machine Learning, 1263-1272. arXiv:1704.01212.
Xu, K., Hu, W., Leskovec, J., & Jegelka, S. (2019). How powerful are graph neural networks? Proceedings of the 7th International Conference on Learning Representations (ICLR). arXiv:1810.00826.
Duvenaud, D., Maclaurin, D., Aguilera-Iparraguirre, J., Gomez-Bombarelli, R., Hirzel, T., Aspuru-Guzik, A., & Adams, R. P. (2015). Convolutional networks on graphs for learning molecular fingerprints. Advances in Neural Information Processing Systems, 28, 2224-2232.
Ullmann, J. R. (1976). An algorithm for subgraph isomorphism. Journal of the ACM, 23(1), 31-42. doi:10.1145/321921.321925
Cordella, L. P., Foggia, P., Sansone, C., & Vento, M. (2004). A (sub)graph isomorphism algorithm for matching large graphs. IEEE Transactions on Pattern Analysis and Machine Intelligence, 26(10), 1367-1372. doi:10.1109/TPAMI.2004.75
Willett, P., Barnard, J. M., & Downs, G. M. (1998). Chemical similarity searching. Journal of Chemical Information and Computer Sciences, 38(6), 983-996. doi:10.1021/ci9800211
Segler, M. H. S., Preuss, M., & Waller, M. P. (2018). Planning chemical syntheses with deep neural networks and symbolic AI. Nature, 555, 604-610. doi:10.1038/nature25978
Warr, W. A. (2010). Tautomerism in chemical information management systems. Journal of Computer-Aided Molecular Design, 24(6-7), 497-520. doi:10.1007/s10822-010-9338-4
Babai, L. (2016). Graph isomorphism in quasipolynomial time. Proceedings of the 48th Annual ACM Symposium on Theory of Computing (STOC), 684-697. doi:10.1145/2897518.2897542